MetaHipMer¶
Background¶
MetaHipMer (MHM) is a de novo metagenome short-read assembler. The recent version of MHM is written completely in UPC++ and can run on both single node servers and large scale supercomputers. MHM can scale upto thousands of nodes to coassemble terabase-sized metagenomes. Metagenome assembly involves converting sets of short, overlapping, and error prone DNA segments from environmental samples into the accurate representation of the underlying microbiomes’s genomes. MetaHipMer uses algorithmic motifs which are uncommon across other science domains like large scale distributed hash tables, graph traversal, sequence alignment and sequence extension using hash table walks. All of these motifs are not typical for GPU offloading. Most of the modules of MetaHipMer are communication bound and do not use floating point operations. Most of the computations are integer heavy.
Alignment Phase¶
MetaHipMer's alignment phase involves aligning one sequence with a set of other sequences. Typically, before the alignment is performed, sequences to be aligned are gathered from different nodes and then alignment computations are performed local to each node. At scale, the communication portion of this phase is dominant but for smaller number of nodes the alignment kernel takes up large chunk of alignment time. Sequence alignment is performed using a popular dynamic programming algorithm known as smith-waterman alignment algorithm. We started off by offloading this algorithm's kernel to GPUs. Existing GPU implementations for Smith Waterman algorithm were not suitable for our use case so we had to implement one that worked for our use case. Below are some details of the process we used to implement this.
Smith Waterman Alignment Algorithm¶
Smith Waterman (SW) Algorithm is a dynamic programming algorithms that are used to compute all possible local alignments between two biological sequences and then find the optimal alignment between two. This requires filling up a scoring matrix which has a rather unique memory access pattern. At a given time, only the elements across the minor diagonal can be computed, this induces a memory access pattern along the minor diagonal and introduces dependencies at each iteration. Below figure shows that the matrix is filled up in three stages, the first stage (shaded in yellow) is when the size of anti-diagonal is increasing i.e. the number of cells that can be computed in parallel are increasing. Second stage is (shaded in orange) when the diagonal size remains fixed and the number of elements that can be computed concurrently remain fixed and finally third stage is (shaded in red) where the number of elements that can be computed in parallel at a given time decrease with each iteration. 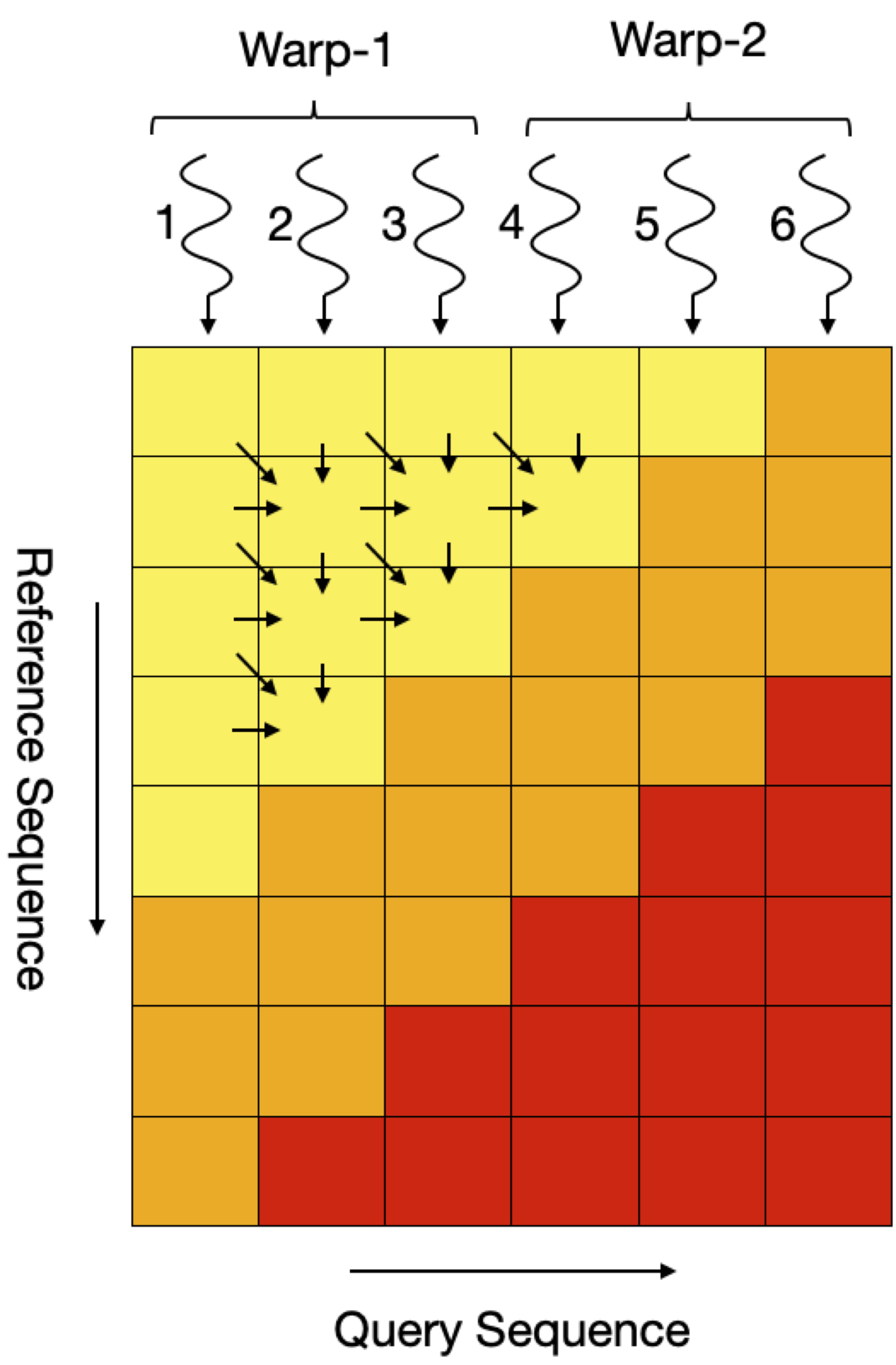
Challenges on GPU porting¶
- Memory accesses along the anti-diagonal of a matrix means that two elements that can be computed in parallel at a given time would be placed about 200 bytes apart. This is not an ideal scenerio for a GPU because memory accesses on GPUs are processed in transactions and if elements to be computed are placed more than cache line size apart, this may lead to multiple memory transactions which reduce bandwidth utilization.
- Changing parallelism through out the life of algorithm and the dependencies in each iteration also pose an additional challenge for GPU computing.
Implementation¶
In the CPU only code, alignment phase processes each alignment on the run but for GPU we batch up the alignments and then offload them to GPU once the number of alignments are large enough to take advantage of GPU. One alignment is performed per CUDA block, within each CUDA block there is a more find grained implementation which is discussed in detail below. Our implementation of Smith-Waterman algorithm is named ADEPT.
Tracking Inter-thread dependencies¶
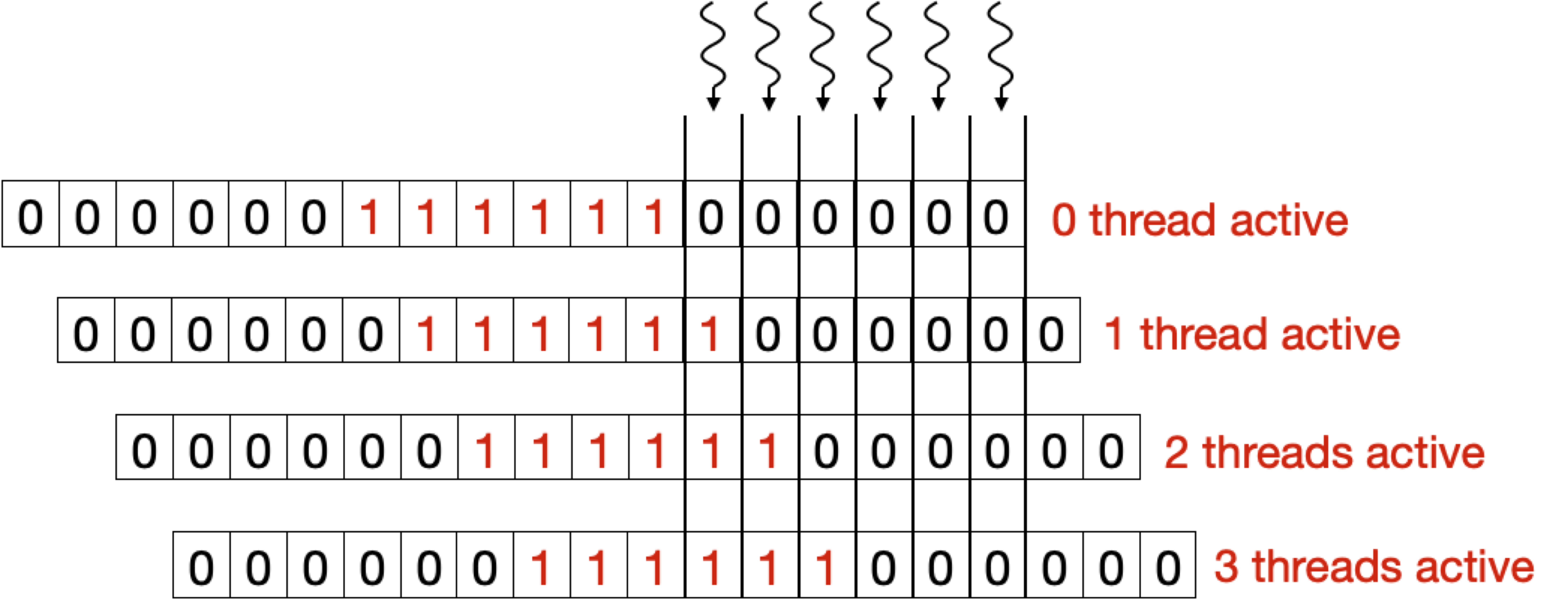
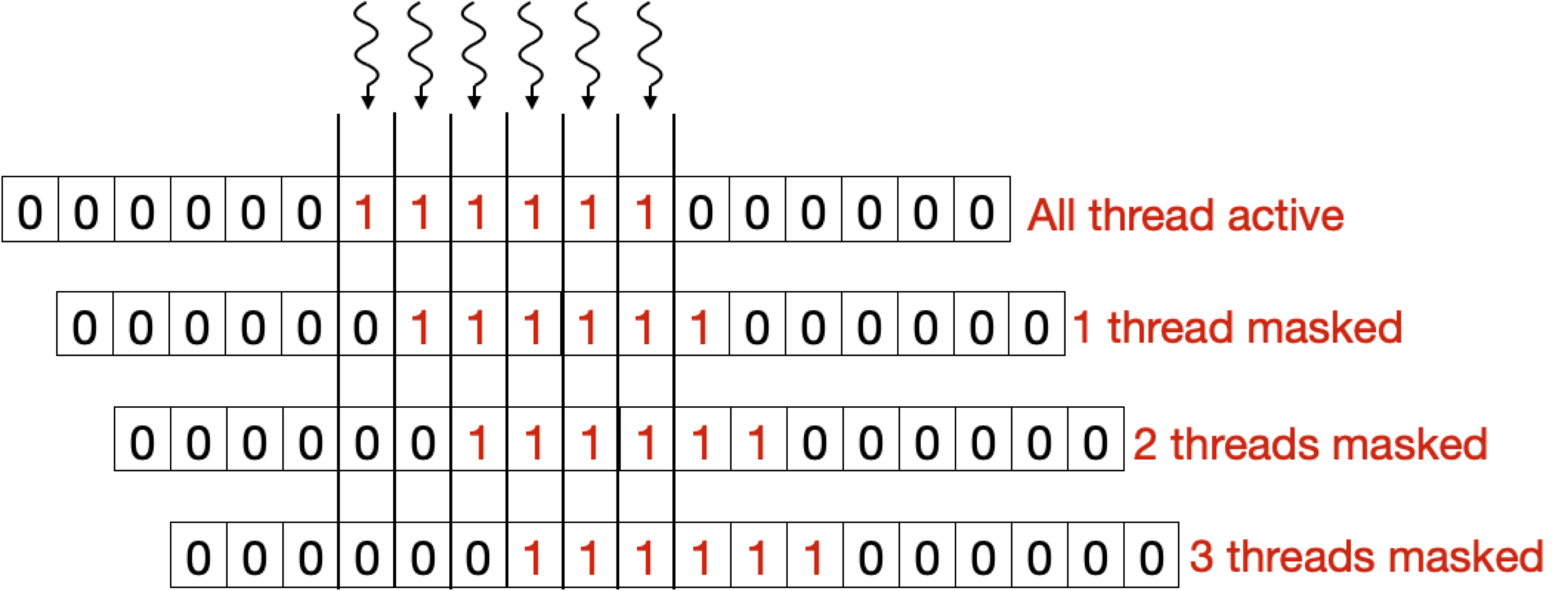
The maximum number of cells that can be calculated in parallel at any given time is equal to the length of the shorter of the two sequences. This poses the challenge of keeping track of dependencies for different cells and masking out the threads for which dependencies are not ready. To tackle this problem, we introduce a Binary Masking Array (BMA) for masking out threads in each iteration for which dependencies are not ready.
Below two figure shows the BMA array for a query of length 6. Here, each thread keeps track of an element in BMA. After each iteration, if the algorithm is in the yellow region (see above Figure), the array shifts to right, activating one more thread. While in the orange region the array stops shifting to keep all the threads activated, when it enters the red region the array starts shifting again but this time each shift deactivates a thread.
Dynamic Programming Table storage and Memory Issues¶
To compute the highest scoring cell a pass over the complete matrix is required. Since the total number of cells that are computed in the matrix are n∗m (lengths of two sequenes), if we use 2 bytes to store each cell, storing the complete dynamic programming table in memory requires N∗(2∗mn) bytes. Where N is the size of a batch. This yields a total global memory requirement of several hundred GBs for a million alignments, and even top of the line GPUs have global memory of only a few GBs.
Apart from the storage size of the dynamic programming table, another challenge that occurs often on GPUs is that of non-coalesced global memory accesses. The Global memory accessed by threads of a CUDA warp is bundled into the minimum number of cache loads, where L1 cache line size is 128 bytes. Thus if the two threads are accessing a memory location that is more than 128 bytes apart, the memory accesses will be un-coalesced.
To compute a given anti-diagonal only the two recent most anti-diagonals are required.Thus, we can effectively discard the scoring matrix beyond the two most recent diagonals. Since this requires storing only a portion of the matrix, this can be done inside thread registers thus avoiding the problem of non-coalesced memory accesses.
Efficient Inter thread communication¶
To compute a cell, each thread needs data from its neighboring threads that was computed in the last iteration. For this inter-thread transfer we explored two methods of data sharing between threads i.e. communication using shared memory and register-to-register memory transfer.
CUDA’s warp shuffle intrinsics allow threads to perform direct register-to-register data exchange without performing any memory loads and stores, while use of shared memory involves going through the on-chip shared memory. Due to much faster performance we opted for the register-to-register data exchange method.
However, register-to-register transfers are only allowed among the non-predicated threads of the same warp. This introduces several edge cases, for example in the CUDA platform where a warp is 32 threads wide, a communication between thread (32∗q)−1 and 32∗q (where q>0) would not be possible through register-to-register transfers because these do not belong to same warp. For such edge cases we use shared memory.
Using the above method provides fast inter-thread communication along with freeing up significant amount of shared memory, which helps improve GPU utilization and also helps avoid shared memory bank conflicts. A bank conflict occurs when multiple threads access same bank of shared memory, this enforces sequential access to that portion of memory and results in performance degradation.
Driver function for easy integeration in MetaHipMer¶
ADEPT contains a driver component which manages all the communication, load balancing and batch size determination for the GPU kernels while keeping the developer oblivious of these intricacies.
ADEPT’s driver gathers hardware information about all the GPUs installed on a node and then divides the work equally among them. A separate context is created for each GPU where a unique CPU thread is assigned to a particular GPU. This CPU thread divides the total computational load into smaller batch sizes depending on the memory capacity of the GPU assigned to it. Each batch is then prepared and packed into a data structure which is then passed to a GPU kernel call. Using CUDA streams, the GPU kernel call and the data packing stage are overlapped so that CPU and GPU work can be carried out in parallel. An overview of ADEPT’s design can be seen in below figure: 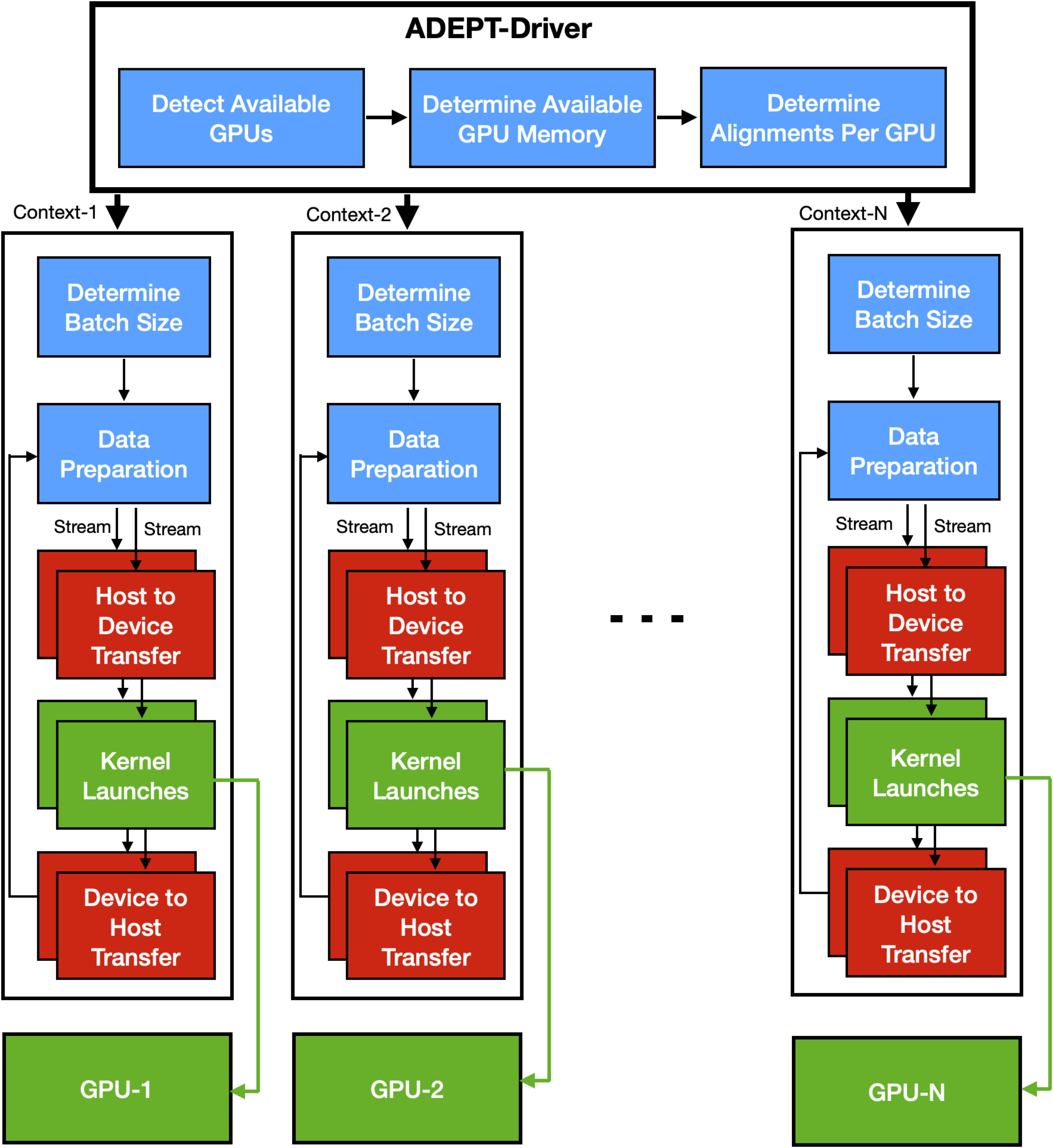
Comparison with similar SIMD methods¶
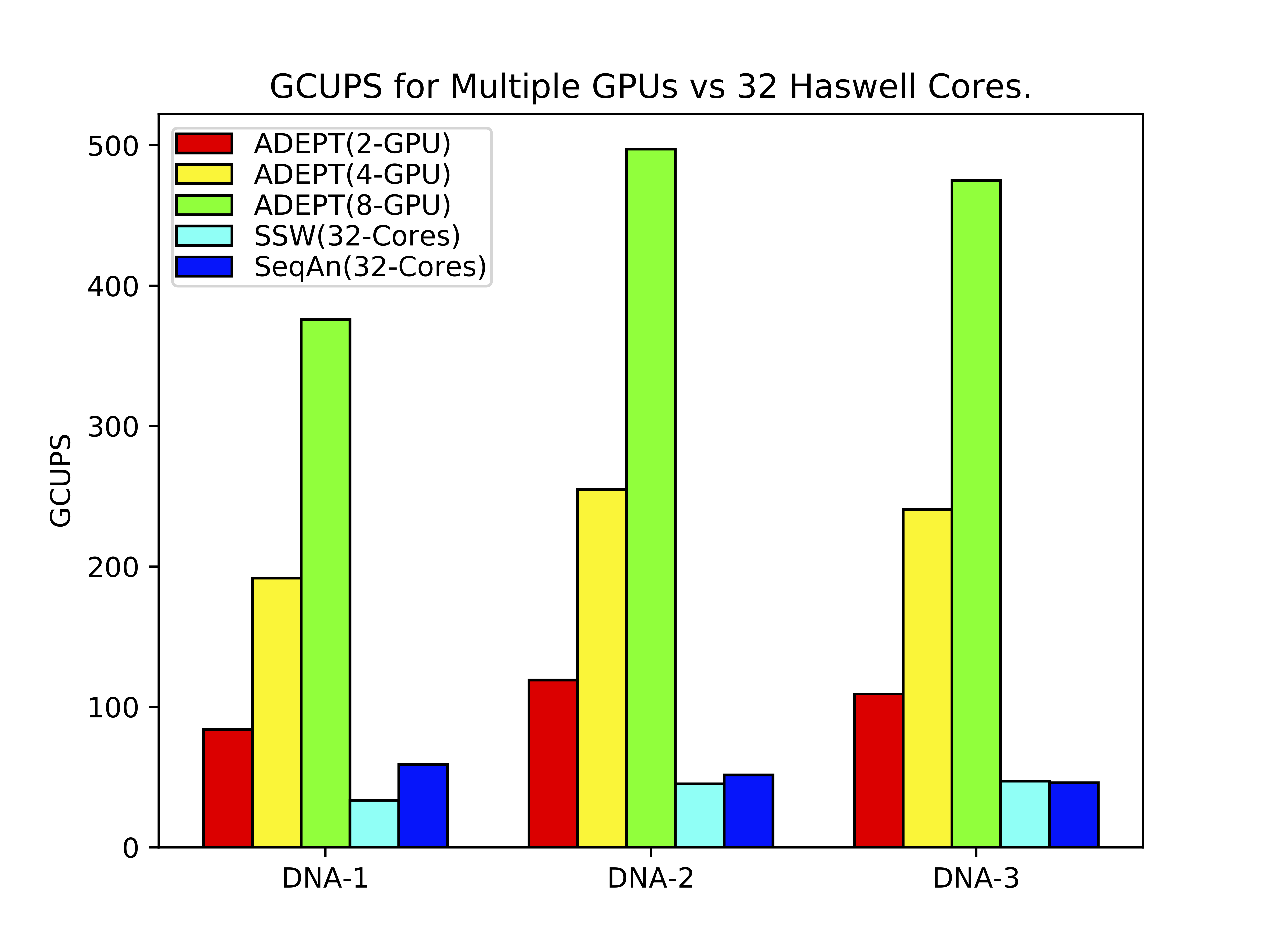
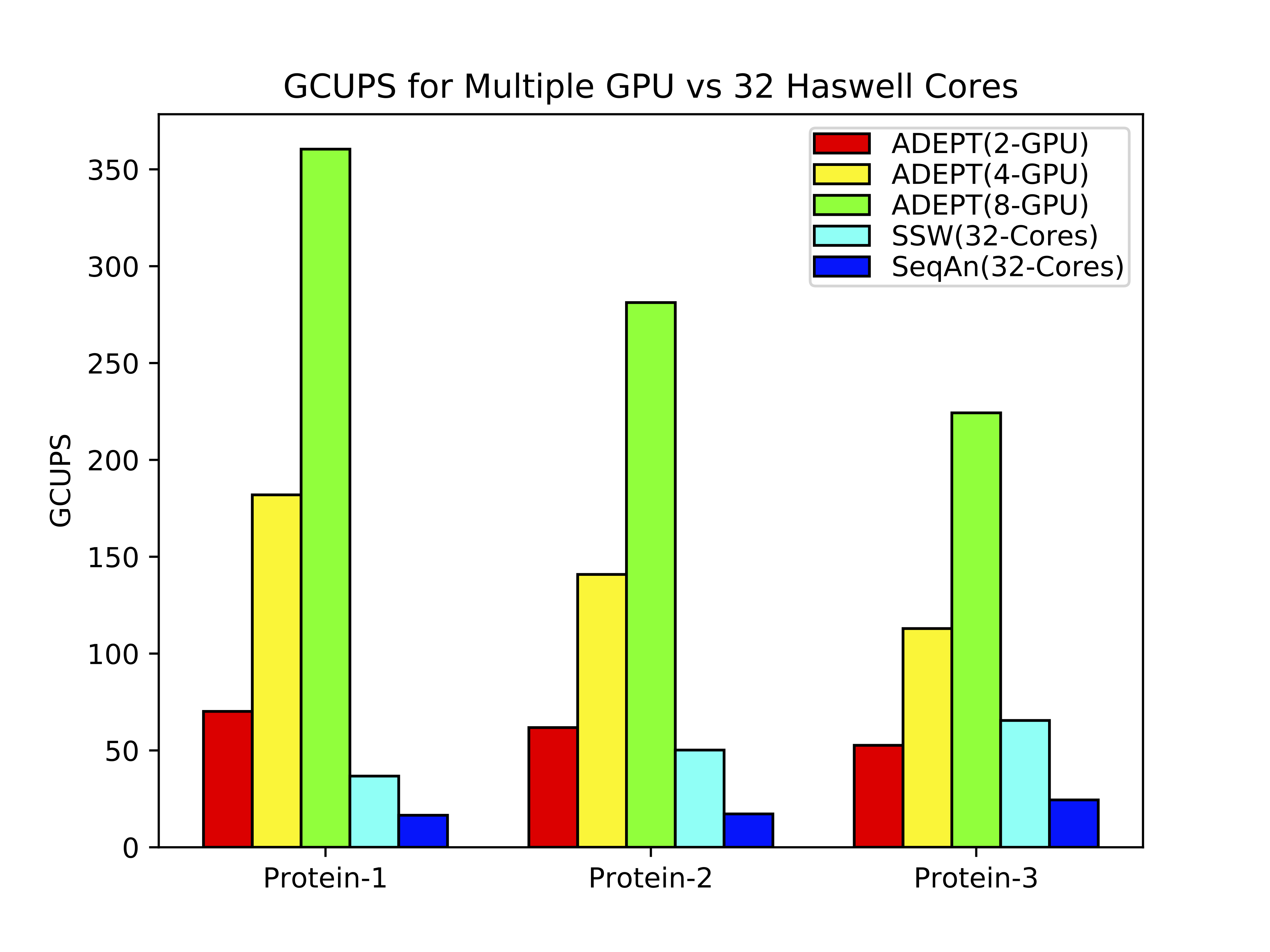
ADEPT implementation was compared against existing SIMD optimized CPU implementations of Smith-Waterman algorithm. For this study we used representative DNA and protein datasets of varying lengths. In the below figure it can be observed that in a node to node comparison (Cori Haswell Node vs Cori GPU node) ADEPT out performs both the CPU optimized methods (SSW and SeQan) by about 10x.
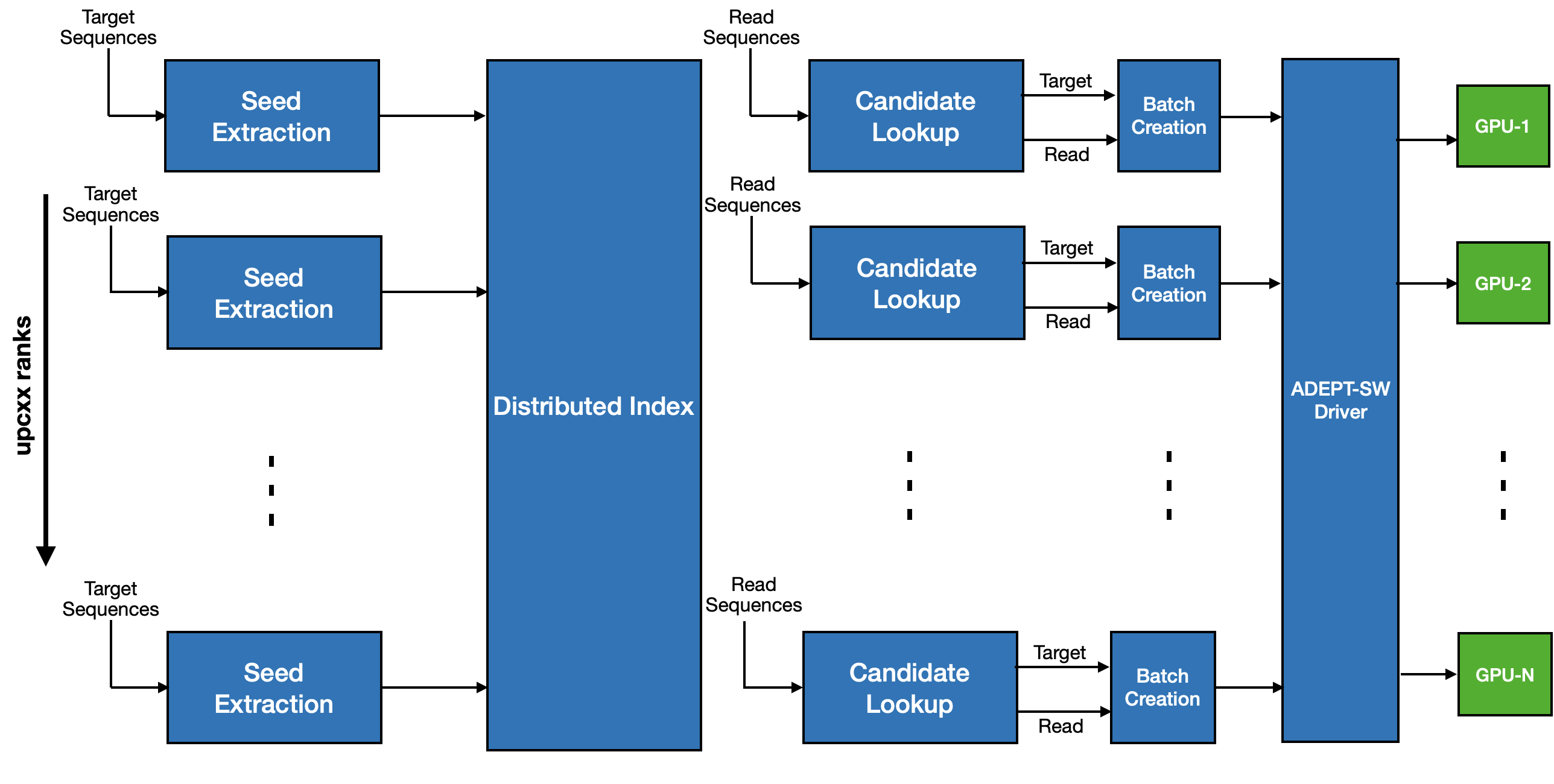
Integeration in MetaHipMer software pipeline¶
Using ADEPT's driver function integeration in MetaHipMer's alignment phase became quite straight forward. As shown in below figure, the sequences that need to aligned are batched up and when the batch size is large enough a call to ADEPT driver is made which maps the calling rank to a GPU and launches the kernel. NVIDIA's Multi Process Service (running in background) can enable execution of multiple kernels on same GPU given the resources are available.
Below figure shows performance comparison of MetaHipMer with and without ADEPT. It can be seen that alignment kernel speeds up considerably when offloaded to GPUs. 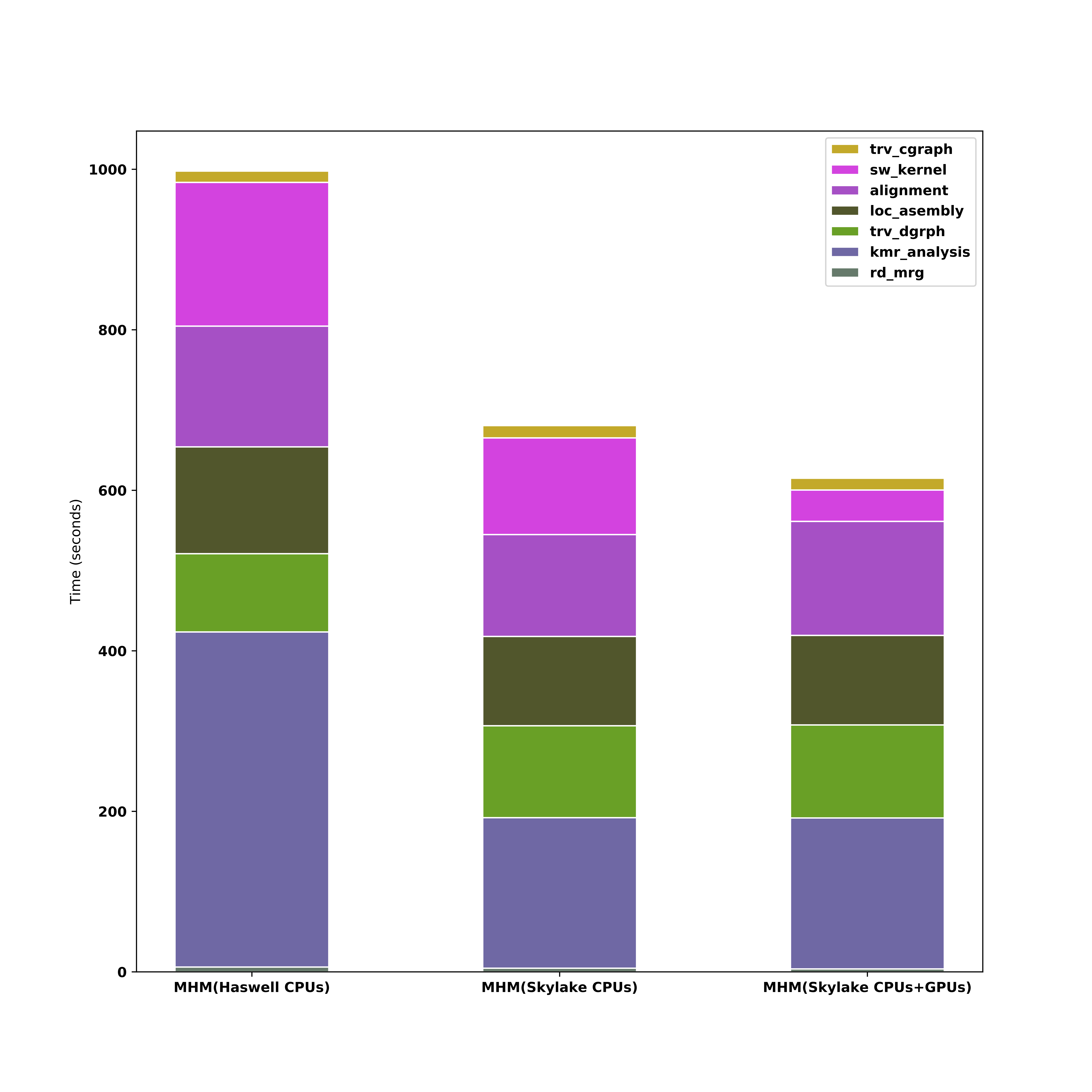
Lessons Learned¶
- Bioinformatics problems are irregular, sparse and do not perform any floating point computations which makes them a poor fit for GPUs. However, as demonstrated in the above case, with the help of low level optimization and significant restructuring of underlying problems advantage can be gained with GPUs.
- In case of an application that is not a good fit for GPU, start of by separating out the most GPU amenable component and working on it independent of the whole application. This allows for flexiblity in trying new and different things.
- Once significant speed up has been achieved on stand alone component, using a suitable interface it can be placed back into the complete application.
- There are more than one ways of inter-thread communication in GPUs, if the problem is sparse then its best to start from the lowest spectrum of memory hierarchy i.e. using registers for inter thread communications. But for some resource hungry problems this might not yield the best results.
- For achieving more concurrency on GPU and increasing GPU utilization one GPU can be mapped to multiple ranks on a node. Such a strategy would allow Multi Process Service to schedule multiple kernels from different ranks on same GPU at a given time thus making better use of device resources.
References¶
- Awan, Muaaz G., et al. "ADEPT: a domain independent sequence alignment strategy for gpu architectures." BMC bioinformatics 21.1 (2020): 1-29.
- Hofmeyr, Steven, et al. "Terabase-scale metagenome coassembly with metahipmer." Scientific reports 10.1 (2020): 1-11.
- Georganas, Evangelos, et al. "Extreme scale de novo metagenome assembly." SC18: International Conference for High Performance Computing, Networking, Storage and Analysis. IEEE, 2018.