VASP¶
VASP is a package for performing ab initio quantum-mechanical molecular dynamics (MD) using pseudopotentials and a plane wave basis set. The approach implemented in VASP is based on a finite-temperature local-density approximation (with the free energy as variational quantity) and an exact evaluation of the instantaneous electronic ground state at each MD step using efficient matrix diagonalization schemes and an efficient Pulay mixing scheme.
Availability and Supported Architectures at NERSC¶
VASP is available at NERSC as a provided support level package for users with an active VASP license.
Gaining Access to VASP Binaries¶
To gain access to the VASP binaries at NERSC through an existing VASP license, please fill out the VASP License Confirmation Request. You can also access this form at NERSC Help Desk (Open Request -> VASP License Confirmation Request).
Note
If your VASP license was purchased from VASP Software GmbH, the license owner (usually your PI) must register you under his/her license at the VASP Portal before you fill out the form.
It may take several business days from when the form is submitted to when access to NERSC-provided VASP binaries is granted.
When your VASP license is confirmed, NERSC will add you to a unix file group: vasp5 for VASP 5, vasp6 for VASP 6 up through VASP 6.4.3, and vasp65 for VASP 6.5 and later. You can check if you have VASP access at NERSC via the groups command. If you are in the vasp5 file group, then you can access VASP 5 binaries provided at NERSC. If you are in the vasp6 file group, then you can access VASP 6 binaries up through VASP version 6.4.3. If you are in the vasp65 group then you can access VASP 6 binaries up through VASP 6.6.
VASP 6.x supports GPU execution.
Versions Supported¶
| Perlmutter GPU | Perlmutter CPU |
|---|---|
| 6.x | 5.4.4, 6.x |
Use the module avail vasp command to see a full list of available sub-versions.
Application Information, Documentation, and Support¶
See the developers page for information about VASP, including links to documentation, workshops, tutorials, and other information. Instructions for building the code and preparing input files can be found in the VASP Online Manual. For troubleshooting, see our troubleshooting guide for frequently asked questions and additional links to support pages.
Using VASP at NERSC¶
We provide multiple VASP builds for users. Use the module avail vasp command to see which versions are available and module load vasp/<version> to load the environment. For example, these are the available modules (as of 06/12/2026),
perlmutter$ module avail vasp
-------------------------------- NERSC Modules ---------------------------------
vasp-tpc/5.4.4-cpu (D) vasp/6.4.2-cpu vasp/6.6.0-cpu
vasp-tpc/6.4.2-cpu vasp/6.4.2-gpu (g) vasp/6.6.0-gpu (g)
vasp-tpc/6.4.2-gpu (g) vasp/6.4.3-cpu
vasp/5.4.4-cpu (D) vasp/6.4.3-gpu (g)
Where:
g: built for GPU
D: Default Module
The "cpu" and "gpu" version strings indicate builds which target Perlmutter's CPU and GPU nodes, respectively.
The modules with "6.x.y" in their version strings are official releases of hybrid MPI+OpenMP VASP, which are available to the users who have VASP 6 licenses. The "vasp-tpc" (tpc stands for "Third Party Codes") modules are the custom builds incorporating commonly used third party contributed codes; these may include Wannier90, DFT-D4, VTST from University of Texas at Austin (available in both CPU and GPU builds); and LIBXC, BEEF, and VASPSol (available only in CPU builds).
The current default on Perlmutter is vasp/5.4.4-cpu (VASP 5.4.4 with the latest patches), and you can access it by
perlmutter$ module load vasp
To use a non-default module, provide the full module name,
perlmutter$ module load vasp/6.4.3-cpu
The module show command shows the effect VASP modules have on your environment, e.g.
----------------------------------------------------------------------------------------------------
/opt/nersc/pe/modulefiles/vasp/5.4.4-cpu.lua:
----------------------------------------------------------------------------------------------------
depends_on("cpu")
whatis("The Vienna Ab initio Simulation Package (VASP) is a computer program for atomic scale materials modelling.")
help([[==========================================================================
Name: vasp
Version: 5.4.4-cpu
URL: https://www.vasp.at/
Description: The Vienna Ab initio Simulation Package (VASP) is a computer program for atomic scale materials modelling.
==========================================================================
VASP is a package for performing ab initio quantum-mechanical molecular
dynamics (MD) using pseudopotentials and a plane wave basis set.
VASP modules are available only for the NERSC users who have an existing VASP license.
To gain access to the VASP binaries at NERSC through an existing VASP
license, please fill out the VASP License Confirmation Request form at
https://help.nersc.gov (Open Request -> VASP License Confirmation Request).
References:
- https://docs.nersc.gov/applications/vasp/
- https://www.vasp.at/
]])
setenv("PSEUDOPOTENTIAL_DIR","/global/common/software/nersc9/vasp/dependencies/pseudopotentials")
setenv("VDW_KERNEL_DIR","/global/common/software/nersc9/vasp/dependencies/vdw_kernel")
setenv("MAKEFILE_INCLUDE_PATH","/global/common/software/nersc9/vasp/vasp/5.4.4-cpu")
setenv("NO_STOP_MESSAGE","1")
setenv("MPICH_NO_BUFFER_ALIAS_CHECK","1")
setenv("OMP_NUM_THREADS","1")
prepend_path("PATH","/global/common/software/nersc9/vasp/dependencies/vtstscripts-1033")
prepend_path("PATH","/global/common/software/nersc9/vasp/dependencies/bader")
prepend_path("PATH","/global/common/software/nersc9/vasp/vasp/5.4.4-cpu/bin")
This vasp module adds the path to the VASP binaries to your search path and sets a few environment variables, where PSEUDOPOTENTIAL_DIR and VDW_KERNEL_DIR are defined for the locations of the pseudopotential files and the vdw_kernel.bindat file used in dispersion calculations.
VASP binaries¶
Each VASP module provides three different binaries:
vasp_gam- gamma-point-only buildvasp_ncl- non-collinear spinvasp_std- the standard k-point binary
One must choose the appropriate binary for the corresponding job.
Sample Job Scripts¶
To run batch jobs, prepare a job script (see samples below), and submit it to the batch system with the sbatch command, e.g. for job script named run.slurm,
nersc$ sbatch run.slurm
Please check the Queue Policy page for the available QOS settings and their resource limits.
Perlmutter GPUs¶
Sample job script for running VASP 6 on Perlmutter GPU nodes
#!/bin/bash
#SBATCH -A <your account name> # e.g., m1111
#SBATCH -C gpu
#SBATCH -q regular
#SBATCH -N 2
#SBATCH -t 01:00:00
#SBATCH -J vasp_job
#SBATCH -o %x-%j.out
#SBATCH -e %x-%j.err
module load vasp/6.4.3-gpu
# One can use up to 16 OpenMP threads-per-MPI-rank when using
# 4 GPUs-per-node.
export OMP_NUM_THREADS=1
export OMP_PLACES=threads
export OMP_PROC_BIND=spread
# Run 8 MPI ranks (-n 8) with 8 GPUs (-G 8) on 2 nodes:
# The number of MPI ranks MUST match the number of GPUs.
# The number of GPUs-per-node may not exceed 4.
# Set -c ("--cpus-per-task") = 32 for the ideal MPI rank spacing.
srun -n 8 -c 32 -G 8 --cpu-bind=cores --gpu-bind=none vasp_std
Perlmutter CPUs¶
Sample job script for running VASP 5 on Perlmutter CPU nodes
#!/bin/bash
#SBATCH -A <your account name> # e.g., m1111
#SBATCH -C cpu
#SBATCH -q regular
#SBATCH -N 2
#SBATCH -t 01:00:00
#SBATCH -J vasp_job
#SBATCH -o %x-%j.out
#SBATCH -e %x-%j.err
# Default version loaded: vasp/5.4.4-cpu
module load vasp
# Run with (-n) 256 total MPI ranks
# 128-MPI-ranks-per-node is maximum on Perlmutter CPU
# Set -c ("--cpus-per-task") = 2
# to space processes two "logical cores" apart
srun -n 256 -c 2 --cpu-bind=cores vasp_std
Sample job script for running VASP 6 on Perlmutter CPU nodes
#!/bin/bash
#SBATCH -A <your account name> # e.g., m1111
#SBATCH -C cpu
#SBATCH -q regular
#SBATCH -N 2
#SBATCH -t 01:00:00
#SBATCH -J vasp_job
#SBATCH -o %x-%j.out
#SBATCH -e %x-%j.err
module load vasp/6.4.3-cpu
# Always provide OpenMP settings when running VASP 6
export OMP_NUM_THREADS=2
export OMP_PLACES=threads
export OMP_PROC_BIND=spread
# Run with (-n) 128 total MPI ranks:
# 64 MPI-ranks-per-node
# 2 OpenMP threads-per-MPI-rank
# Set -c ("--cpus-per-task") = 2 x (OMP_NUM_THREADS) = 4
# to space processes two "logical cores" apart
srun -n 128 -c 4 --cpu-bind=cores vasp_std
Running interactively¶
To run VASP interactively, request a batch session using salloc.
Tips
- The interactive QOS allocates the requested nodes immediately or cancels your job in about 5 minutes (when no nodes are available). See the Queue Policy page for more info.
- Test your job using the interactive QOS before submitting a long running job.
Long running VASP jobs¶
For long VASP jobs (e.g., > 48 hours), you can use the variable-time job script, which allows you to run jobs with any length. Variable-time jobs split a long running job into multiple chunks, so it requires the application to be able to restart from where it left off. Note that not all VASP computations are restartable, but e.g., RPA; long running atomic relaxations and MD simulations are good use cases of the variable-time job script.
Sample variable-time job script
#!/bin/bash
#SBATCH -N 1
#SBATCH -C cpu
#SBATCH -J vasp_job
#SBATCH -o %x-%j.out
#SBATCH -e %x-%j.err
#SBATCH --qos=debug_preempt
#SBATCH --comment=0:45:00
#SBATCH --time=0:05:00
#SBATCH --time-min=0:05:00
#SBATCH --signal=B:USR1@60
#SBATCH --requeue
#SBATCH --open-mode=append
## Notes on parameters above:
##
## '--qos=XX' can be set to any of several QoS to which the user has access, which
## may include: regular, debug, shared, preempt, debug-preempt, premium,
## overrun, or shared-overrun.
## '--comment=XX' is the total time that SUM of restarts can run (can be VERY LARGE).
## '--time=XX' is the maximum time that individual restart can run. This MUST fit
## inside the time limit for the QOS that you want to use
## (see https://docs.nersc.gov/jobs/policy/ for details).
## '--time-min=XX' is the minimum time that job can run before being preempted (using this
## option can make it easier for Slurm to fit into the queue, possibly allowing
## job to start sooner). Omit this parameter unless running in either
## --qos=preempt or --qos=debug_preempt.
## '--signal=B:USR1@60' sends signal to begin checkpointing @XX seconds before end-of-job
## (set this large enough to have enough time to write checkpoint file(s)
## before time limit is reached; 60 seconds is usually enough).
## '--requeue' specifies job is elegible for requeue in case of preemption.
## '--open-mode=append' appends contents of to the end of standard output and standard
## error files with successive requeues.
# Remove STOPCAR file so job isn't blocked
if [ -f "STOPCAR" ]; then
rm STOPCAR
fi
# Select VASP module of choice
module load vasp/5.4.4-cpu
# srun must execute in background and catch signal on wait command
# so ampersand ('&') is REQUIRED here
srun -n 128 -c 2 --cpu_bind=cores vasp_std &
# Put any commands that need to run to continue the next job (fragment) here
ckpt_vasp() {
set -x
restarts=`squeue -h -O restartcnt -j $SLURM_JOB_ID`
echo checkpointing the ${restarts}-th job
# Trim space from restarts variable for inclusion into filenames
restarts_num=$(echo $restarts | sed -e 's/^[ \t]*//')
echo "Restart number: ==${restarts_num}=="
# Terminate VASP at the next electronic step
echo LABORT = .TRUE. >STOPCAR
# Wait until VASP completes current step, then write WAVECAR file and quit
srun_pid=$(ps -fle | grep srun | head -1 | awk '{print $4}')
echo srun pid is $srun_pid
wait $srun_pid
# Copy CONTCAR to POSCAR and back up data from current run in each folder
folder="checkpt-$SLURM_JOB_ID-${restarts_num}"
mkdir $folder
echo "In directory $folder"
cp -p CONTCAR POSCAR
cp -p CONTCAR "$folder/POSCAR"
echo "CONTCAR copied."
cp -p OUTCAR "$folder/OUTCAR-${restarts_num}"
echo "OUTCAR copied."
cp -p OSZICAR "$folder/OSZICAR-${restarts_num}"
echo "OSZICAR copied."
# Back up the vasprun.xml file in the parent folder
cp -p vasprun.xml "vasprun-${restarts_num}.xml"
echo "vasprun.xml copied."
set +x
}
ckpt_command=ckpt_vasp
# The 'max_timelimit' is max time per individual job, in seconds
# This line MUST be included !!!
# This MUST match the value set for '#SBATCH --time=' above !!!
max_timelimit=300
# The 'ckpt_overhead' is the time reserved to perform the checkpoint step, in seconds
# This MUST fit within the max_timelimit !!!
# This should match the value set for '--signal=B:USR1@XX' above
ckpt_overhead=60
# Requeue the job if remaining time > 0
. /global/common/sw/cray/cnl7/haswell/nersc_cr/19.10/etc/env_setup.sh
requeue_job func_trap USR1
wait
Running multiple VASP jobs simultaneously¶
For running many similar VASP jobs, it may be beneficial to bundle them inside a single job script, as described in Running Jobs.
However, the maximum number of jobs you should bundle in a job script is limited, ideally not exceeding ten. This is because the batch system, Slurm (as it's implemented currently) is serving tens of thousands of other jobs on the system at the same time as yours, and compounding srun commands can occupy a great deal of Slurm's resources.
Note
Be aware that running too many VASP jobs at once may overwhelm the file system where your job is running. Please do not run jobs in your global home directory.
If you want to run many more similar VASP jobs simultaneously, we recommend using the Fireworks workflow management tool.
Troubleshooting VASP¶
Solutions to frequently encountered issues¶
General Tips
-
Use the most recent version of VASP allowed by your license.
-
Check your job script,
INCAR,POSCAR, andKPOINTSfiles for typos or other input mistakes before running. -
Check the
OUTCAR, standard out, and standard error files to verify that inputs were processed correctly and for error messages. -
Avoid setting environment variables and loading modules in your dotfiles (e.g.
.bashrcresiding in your$HOMEdirectory) when using VASP. User-customized environments may conflict with the environment set by a VASP module and may cause VASP to behave in an unexpected manner. -
Invoke VASP with the flag
stdbuf --output=Lto ensure that error messages are not trapped in the output buffer if the job fails, e.g.srun -n 4 -c 64 --cpu-bind=cores stdbuf --output=L vasp_std -
If you suspect that your job is running abnormally, then try logging into the compute node where your job is running using the
ssh nid<XXXX>command (where<XXXX>corresponds to the node ID). Once logged in, use thetopandnvidia-smicommands to monitor CPU and GPU activity, respectively.
My VASP job just sits in the queue and does not start.
-
Nodes may be unavailable due to a scheduled maintenance or to an unexpected outage or to a large node reservation. In this case the
squeuecommand will typically display a message similar to the following:Your job will be held in the queue until the nodes become available. See the NERSC Center Status page for details.elvis@perlmutter:login28:~> squeue -u elvis JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON) 24224393 gpu_ss11 vasp_job elvis PD 0:00 1 (ReqNodeNotAvail, UnavailableNodes:nid[001140-008864]) -
Consider whether the amount of resources (nodes, wall time) that you requested are appropriate for your job, and decrease the size of the request if possible. As a general rule, the most reliable way to accelerate your job's priority in the queue is to reduce the time limit as this gives Slurm the flexibility to fit your job in gaps between other jobs.
My VASP job immediately crashes with the message: <vasp_executable>: No such file or directory:
Example:
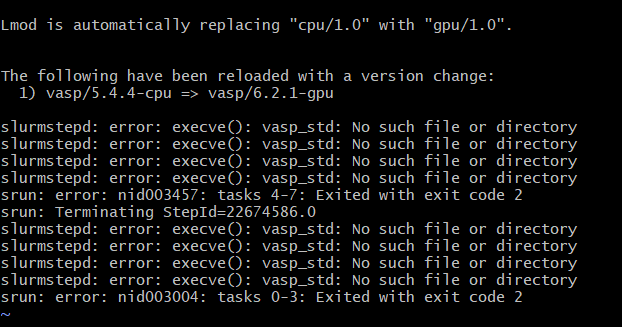
VASP binaries are permission-locked to file groups and are not visible to users outside of the file groups. This message appears when you attempt to run a VASP executable without being in the corresponding file group. Take the following steps:
- Once logged in to NERSC, execute the
groupscommand to show the file groups to which you belong. You must be in thevasp5group to access VASP 5 binaries and in thevasp6group to access VASP 6 binaries. - Fill out the VASP License Confirmation Request form to request access. Once your VASP license has been successfully confirmed, NERSC will add you to the file groups corresponding to your license.
- Check the list of currently available versions of VASP via the command
module avail vasp. The message above can also appear if your job script loaded a VASP module that is no longer available.
My VASP job crashes with an error when trying to read {POSCAR|INCAR|KPOINTS}, but the file appears to be correct.
Example:
ERROR: there must be 1 or 3 items on line 2 of POSCAR
This error can occur when reading input files generated by an external program or by a text editor on a non-unix operating system. Here, the input file contains special (non-human-readable) characters that cause VASP to read the file incorrectly. To test if this is the case:
-
Back up the input file which causes the error, e.g.
perlmutter$ mv POSCAR POSCAR_backup -
Pipe the text of the input file to screen:
perlmutter$ cat POSCAR_backup -
Create a new input file (in this example,
POSCAR) using a unix-based text editor (e.g.vimornano) and paste the text from the preceding step into the new file, and save the new file. -
Run your VASP job using the new input file.
If the preceding steps solve the issue, then possible production solutions include:
-
Check to see if your external input generating program can export files to a unix-compatible format.
-
Use a tool to convert the files to unix format. For example, one can convert DOS files via the
dos2unixutility, e.g.perlmutter$ dos2unix POSCAR
My VASP job runs with settings inconsistent with those set in my INCAR file
This issue has two likely causes:
-
If the
INCARfile was created by a text editor on a non-unix operating system, then the file may contains special (non-human-readable) characters that cause VASP to read the file incorrectly. See preceding entry for how to test and troubleshoot. -
If a section of the
INCARfile is commented incorrectly, the commented-out section may be read as input parameter(s). Always use Python-style comments# like thisto comment out text in yourINCAR.
My VASP job immediately crashes with an error about not being compiled for noncollinear calculations
ERROR: noncollinear calculations require that VASP is compiled without the flag -DNGXhalf and -DNGZhalf
This error occurs when one attempts to run a noncollinear calculation using the vasp_std or vasp_gam executable. To solve, use the vasp_ncl executable instead.
My VASP job runs but produces no output or incomplete output
This type of error can occur when one incorrectly configures how a job interacts with the file system. Here are some items to check:
-
Always run jobs in your
$SCRATCHdirectory, not in$HOME. VASP generates large files which, if run in$HOME, may cause your$HOMEdirectory to exceed the quota. After the quota has been reached, no new files may be written, and your output will be incomplete or missing. -
When running multiple VASP calculations inside of a single job script, perform each calculation in a separate directory. VASP generates identically-named files (
OUTCAR,OSZICAR, ...) for each calculation, and these files will overwrite one another if run in the same directory.
My VASP job hangs or stops making progress
There are various reasons that a VASP job may hang, including:
-
VASP jobs on GPUs may hang due to incorrect binding. Examples of this include:
-
VASP uses NCCL to transfer data between GPUs attached to different MPI ranks. If the Slurm setting
--gpu-bind=single:1is used it will cause only one GPU to be visible to each MPI rank; this inhibits NCCL and can cause GPU jobs to hang. Set--gpu-bind=noneinstead. -
VASP only supports 1:1 MPI-rank-to-GPU mapping. This means that one cannot use multiple MPI ranks per GPU, nor can one use multiple GPUs per MPI rank. Make sure to select no more than four MPI ranks per node and that the number of GPUs is set equal to the number of MPI ranks in your job script.
-
-
Running on hyperthreads, or running multiple processes per physical cores, can cause VASP jobs to hang:
-
On Perlmutter GPU, each node contains 64 physical cores but 128 "logical cores" with hyperthreading. Make sure that you do not run more than four MPI ranks per node, and, if using OpenMP, make sure that the product of MPI ranks per node and OpenMP threads-per-MPI-rank does not exceed 64. The value for the Slurm parameter
-c(--cpus-per-task) should not be set less than 32. -
On Perlmutter CPU, each node contains 128 physical cores but 256 "logical cores" with hyperthreading. Make sure that you do not run more than 128 MPI ranks per node (for a "pure" MPI calculation), or, if using OpenMP, make sure that the product of MPI ranks per node and OpenMP threads per MPI rank does not exceed 128. The value for the Slurm parameter
-c(--cpus-per-task) should never be set less than 2. -
When running VASP 6 executables with OpenMP enabled, set the OpenMP environment variables after loading the VASP module but before invoking
srunto ensure that the correct OpenMP settings are selected. We recommend always setting the OpenMP variables as follows, even when not using OpenMP explicitly:export OMP_NUM_THREADS=1 # Set >1 if using OpenMP export OMP_PLACES=threads export OMP_PROC_BIND=spread
-
-
Some plugins/third-party codes do not run on GPUs and may hang when run with the GPU version of VASP. If you have an issue running a third party code with a VASP GPU module, try running using the corresponding CPU module.
-
Calculations on very small systems may hang if run with too many MPI ranks.
My VASP job fails with the message cxil_map: write error
This error is caused by a known vendor bug and can appear in GPU jobs that use MPI. Although this bug affects multiple applications, this error occurs only sporadically in VASP runs and can be usually solved by rerunning your VASP job.
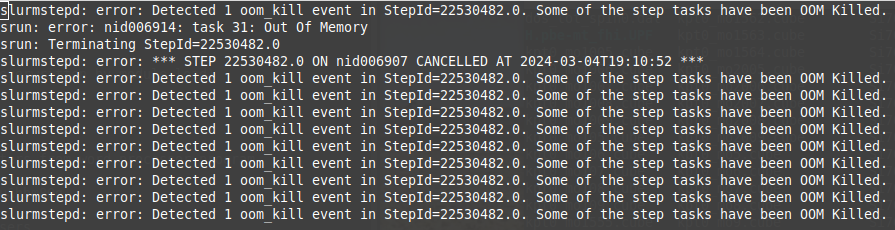
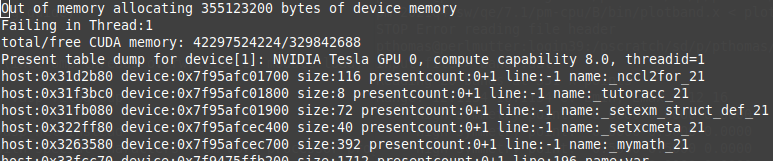
My VASP job runs out of memory
VASP jobs containing many atoms, many k-points, or large FFT grids may fail due to lack of memory resources. Please see the VASP Wiki Not Enough Memory page for an explanation of VASP memory usage. The general strategy to solve out-of-memory errors is to increase the number of nodes or GPUs requested as this increases the pool of available memory.
Sample out-of-memory failure when exceeding host (CPU) memory:
Sample out-of-memory failure when exceeding device (GPU) memory:
To solve out-of-memory errors, try the following steps:
- Check the memory cost table (in
OUTCAR) to make sure that the cost is what you expect. Below is a sample table:
total amount of memory used by VASP MPI-rank0 42743434. kBytes
=======================================================================
base : 30000. kBytes
nonlr-proj: 97297. kBytes
fftplans : 63529. kBytes
grid : 228114. kBytes
one-center: 9953. kBytes
wavefun : 42314541. kBytes
-
Make a "test" run of your job with a short time limit, and examine how the memory cost table changes as you increase the number of nodes. Try increasing the number of nodes while leaving the number of MPI ranks-per-node constant. Alternatively, you may find it helpful to increase the number of nodes while leaving the total number of MPI ranks constant. Increase the node count until the value for "total amount of memory used by VASP MPI-rank0" multiplied by the number of ranks-per-node of your job fits inside the node's memory limit (512 GB and 256 GB for Perlmutter CPU and GPU nodes, respectively).
-
For GPU jobs, one may also be limited by memory on the GPU card itself. The Slurm setting
#SBATCH -C gpumay request either 40 GB or 80 GB A100 GPUs. One can explicitly request only 80 GB A100 GPU nodes by setting#SBATCH -C gpu&hbm80ginstead. Please keep in mind that the 80 GB nodes are a limited commodity and that your queue time may increase significantly with this setting. -
The choice of VASP executable also influences the memory cost, with
vasp_ncl>vasp_std>vasp_gam. Check if your calculation can be completed with an executable which uses less memory. -
K-point parallelization improves performance but also increases memory usage. You can reduce the memory cost by decreasing the value of KPAR in your
INCARfile, with KPAR=1 giving the lowest memory usage.
My VASP job crashes with a '!BUG!' message in my OUTCAR file
Example:
-----------------------------------------------------------------------------
| _ ____ _ _ _____ _ |
| | | | _ \ | | | | / ____| | | |
| | | | |_) | | | | | | | __ | | |
| |_| | _ < | | | | | | |_ | |_| |
| _ | |_) | | |__| | | |__| | _ |
| (_) |____/ \____/ \_____| (_) |
| |
| internal error in: mkpoints_change.F at line: 185 |
| |
| internal ERROR in RE_READ_KPOINTS_RD: the new k-point set for the |
| reduced symmetry case |
| does not contain all original k-points. Try to switch off symmetry |
| |
| If you are not a developer, you should not encounter this problem. |
| Please submit a bug report. |
| |
-----------------------------------------------------------------------------
Take the following steps:
-
Try the work-around suggestions provided by the 'BUG!' message, if any.
-
Check the VASP Known Issues page and search the VASP User Forum to see if this issue has been previously reported.
-
If this issue has not been reported, file a bug report with the VASP developers via the VASP Portal. Be sure to provide the VASP developers with all of the input files needed to reproduce your issue (
INCAR,POSCAR,KPOINTSfiles, along with your job script). -
If the VASP developers provide a patch or recommend a change in the build, contact the NERSC Help Desk for further assistance, and provide NERSC with a link to your post on the VASP User Forum.
My VASP jobs are running but there are problems with convergence in the SCF or relaxation
This type of issue has two common causes:
-
The positions of the atoms are bad, e.g. two atoms are too close to one another (possibly as a consequence of cell volume or symmetry). This usually occurs at the start of the calculation but it can also result from a geometry update step.
-
The algorithm used for the SCF is a poor match to the electronic structure of the molecule or material system.
For issues with electronic relaxation, see the VASP Developers' documentation for the ALGO parameter. For issues with geometry relaxation, see the VASP Developers' page for the IBRION parameter. There is also a developer-provided guide for Troubleshooting Electronic Convergence.
My VASP jobs crash with an error in a library routine
Example:
Error EDDDAV: Call to ZHEGV failed. Returncode = <value>
This message may be preceded by one or more warnings of the type:
WARNING: Sub-Space-Matrix is not hermitian in DAV
See above issue: "My VASP jobs are running but there are problems with convergence in the SCF or relaxation".
I need a VASP build which contains a particular plugin
The standard VASP modules contain plugins that can be enabled by precompiler options (such as Wannier90). Plugins which require modification of the source code of VASP are instead included in the vasp-tpc modules. These include VTST (used for Nudged Elastic Band calculations) and VASPSol (used to model solvation effects). Note that not all plugins are available in GPU builds. If you require a plugin that is not present in one of the vasp-tpc modules, please file a support ticket.
I need a sample makefile.include to build my own custom VASP
Each NERSC VASP module has an environment variable called MAKEFILE_INCLUDE_PATH which points to the location of the 'makefile.include' used to build the module. To see it, execute module show <vasp-module-of-choice>. For example, to find the makefile.include for the vasp-tpc/6.4.2-cpu module:
> module show vasp-tpc/6.4.2-cpu
...
setenv("MAKEFILE_INCLUDE_PATH","/global/common/software/nersc9/vasp/vasp-tpc/6.4.2-cpu")
...
makefile.include at this path has permissions set so that you can copy it to your own folder. Known issues running VASP on NERSC resources¶
Some VASP builds produce incorrect magnetic susceptibility tensors
Incorrect magnetic susceptibility tensors have been observed in both VASP 5.4.4 and 6.x builds on Perlmutter made with the PrgEnv-nvidia programming environment. Builds with the PrgEnv-gnu and PrgEnv-intel environments appear to give correct values. If you encounter this issue and need assistance, please file a support ticket.
For additional help with troubleshooting¶
If you encounter an issue that is not described above or need further assistance, see the VASP users forum for technical help and support-related questions; see also the list of known issues. For help with issues specific to the NERSC module, please file a support ticket. Below are some general guidelines for where to seek assistance in various cases:
When to file a ticket with the VASP developers:
Examples of issues that should be directed to the VASP developers at the VASP Portal include:
-
You encounter a
!BUG!message while running. -
Your job fails due to algorithmic issues, such as SCF convergence or relaxation.
-
You would like advice related to choosing pseudopotentials, INCAR parameters, or program options.
When to file a ticket with the NERSC Help Desk:
Common examples of issues that should be directed to the NERSC Help Desk include:
-
Your job fails with Slurm or MPICH errors.
-
You have a job which previously ran successfully on Perlmutter using a NERSC-provided module but suddenly begins failing.
-
You need to create a job script for a specific purpose, such as a checkpoint/restart run or an automated workflow.
-
You would like advice specific to compiling or running on Perlmutter.
Advice for filing VASP support tickets
When you file a VASP-related ticket at the NERSC Help Desk, your issue will be initially handled by the Consultant on Duty (CoD) who may or may not be a VASP expert. The CoD will either answer the ticket themselves or will route it to a specialist. In general, the more information that you provide in the initial ticket, the easier it is for the CoD to direct your ticket to the NERSC staff that is best equipped to answer your issue. If you would like for NERSC staff to examine your job, please paste the path to the location where your job ran on Perlmutter in the ticket as this is preferable to attaching files. For the following issue categories, see below for some considerations:
-
Runtime issue: this includes problems that you may have when running an individual VASP calculation, such as Slurm errors, segmentation faults, MPICH errors, and CUDA errors.
-
Workflow issue: this includes most problems that do not appear in a single job but which occur when multiple VASP jobs are involved, such as:
-
Using and automated environment to run a collection of VASP jobs.
-
Running many VASP jobs in parallel using a single job script.
-
Checkpointing and restarting VASP.
-
Setting up a workflow containing VASP jobs which depend on other jobs.
-
Setting up jobs with unusual resource requirements, e.g. very long running jobs or jobs requiring a large fraction of NERSC resources.
-
-
Build issue: this includes problems that you may face if you compile your own version of VASP, such as compiler or linker errors, or if your VASP build has runtime issues that do not appear in the NERSC-provided modules.
For runtime issues, answer the following questions when submitting your request:
-
What incorrect behavior do you observe, and what error messages do you receive (if any)?
-
What is the path to the location of your job on Perlmutter?
-
What is your job script?
Answers to the following questions may also help NERSC diagnose your issue:
-
When did the error first appear?
-
Is the error reproducible when run on different nodes?
-
Did you try a different VASP binary or a different module?
-
Is this error specific to CPU or GPU runs, or does it appear in both?
For workflow issues, please answer the following:
-
What is the "big picture" of what you would like to accomplish with the workflow?
-
At which step in the workflow are you experiencing the issue?
-
What is your job script?
For build issues, please answer the following:
-
Do you intend to run your build on CPUs or GPUs?
-
What is the
makefile.includethat you used to build? -
Which programming environment do you have loaded (e.g.
PrgEnv-gnu)? -
Are there third party codes or other VASP plugins that you included?
-
Did you modify the VASP source code prior to building?
Performance Guidance¶
Users are recommended to refer to the paper to run VASP efficiently on Perlmutter. The VASP developers' parallelization page also provides a detailed explanation of how VASP parallelization works along with advice for optimizing parallel performance.
The following tips may also help improve the performance of VASP jobs:
-
Select the GPU builds whenever possible as GPU performance is superior to CPU performance for most calculation types. Pipelines running many small jobs in parallel may perform better on CPU nodes due to latency in moving data between CPU and GPU.
-
GPU builds for VASP 6.4 and later include OpenMP parallelization, so note that:
-
Without using OpenMP, one can only use up to 4 MPI ranks per node (corresponding to 1:1 MPI-to-GPU mapping), leaving 60 CPU cores per node idle. With OpenMP one can exploit these CPU cores by setting
export OMP_NUM_THREADS=<value>with<value>set as large as 16. -
Creating OpenMP threads requires some overhead which ideally should be amortized by the work done by those threads. Most of the computational workload is already handled by the GPUs, so there may or may not be enough work left over for the CPUs to justify creating the OpenMP threads. The optimal number of OpenMP threads for a calculation depends both on the system size and on details of the algorithms used in the calculation.
-
NCORE parallelism is always disabled (i.e. NCORE is set to 1) in GPU calculations.
-
-
For VASP 5 CPU builds, one can improve parallel performance by setting the NCORE parameter in the
INCARfile. The value of NCORE should factorize the number of MPI ranks per node and should ideally be close to the square root of the number of ranks per node. On Perlmutter CPU using 128 ranks per node, NCORE=8 is usually a reasonable setting. -
For VASP 6 CPU builds, one can improve parallel performance either by setting NCORE or by setting the number of OpenMP threads greater than 1, but not both (i.e. setting
OMP_NUM_THREADS>1 forces NCORE to be set to 1). -
For all VASP builds, one can turn on parallelization over k-points by setting KPAR>1 in the
INCARfile. One should choose a value for KPAR which factorizes the total number of MPI ranks and the number of irreducible k-points.Note
K-point parallelization can significantly improve performance but increases the memory cost of the calculation.
-
Users who run large numbers of small jobs may benefit from using one of the workflow tools supported at NERSC.
We strongly encourage users to perform benchmarking tests with various OpenMP settings and with different values of NCORE and KPAR, as applicable, before beginning production calculations.
Building VASP from Source¶
Some users may be interested in building VASP themselves. As an example we outline the process for building the VASP 6.4.2 binaries. First, download vasp6.4.2.tgz from VASP Portal to your cluster and run,
tar -zxvf vasp6.4.2.tgz
cd vasp.6.4.2
e.g. in your home directory, to unpack the archive and navigate into the VASP main directory.
One needs a makefile.include file to build the code; samples are available in the arch directory in the unpacked archive and are also provided in the installation directories of the NERSC modules. Additional makefile.include examples are available from the VASP developers at the wiki page.
For example, the makefile.include file that we used to build the vasp/6.4.2-gpu module is located at,
/global/common/software/nersc9/vasp/vasp/6.4.2-gpu
Execute module show <a vasp module> to find the installation directory of the NERSC module. Copy the sample makefile.include file from the NERSC installation directory to the root directory of your local VASP 6.4.2 build.
Note
makefile.include files are compiler-specific and must be used with the corresponding programming environment at NERSC. Existing NERSC builds use makefile.include files that are only compatible with the PrgEnv-nvidia environment. Other makefile.include files may require loading a different programming environment.
A user may be interested in augmenting the functionality of VASP by activating certain plugins. See the developer's list of plugin options for instructions on how to modify makefile.include for common supported features.
Note
Plugins must be built using the same compilers used to build VASP, or errors and other unexpected behavior may occur at link time or at run time.
The next step is to prepare your environment for building VASP. The NERSC-provided VASP modules were built after preloading the following modules:
Setting up the GPU build environment
module reset
module load gpu
module load PrgEnv-nvidia
module load cray-hdf5 cray-fftw
module load nccl
Note
Some versions of nccl, including nccl/2.19.4 and nccl/2.21.5 do not work correcly with VASP 6.4.x. We recommend loading nccl/2.18.3-cu12 instead.
Note
VASP versions (6.3 and 6.4) which use the OpenACC GPU port require a patch to build with Cray MPICH. See this VASP forum post for details. This issue is fixed in VASP 6.5.0-.
Setting up the CPU build environment
module reset
module load cpu
module load PrgEnv-nvidia
module load cray-hdf5 cray-fftw
module load intel-mixed
Note
As if June 2026, when building with the intel-mixed/2025.3 module, linking to MKL fails with the message -lmkl_pgi_thread: No such file or directory due to a missing library. As a workaround, load intel-mixed/2023.2 instead of the default intel-mixed when linking VASP with MKL.
Once the module environment is ready, run
make std ncl gam
to build all binaries: vasp_std, vasp_gam, and vasp_ncl.
Related Applications¶
User Contributed Information¶
Please help us improve this page
Users are invited to contribute helpful information and corrections through our GitLab repository.